What’s next in molecular modeling of proteins and biological systems after AlphaFold?
And the latest news from CASP15, the Critical Assessment of Structure Prediction competition
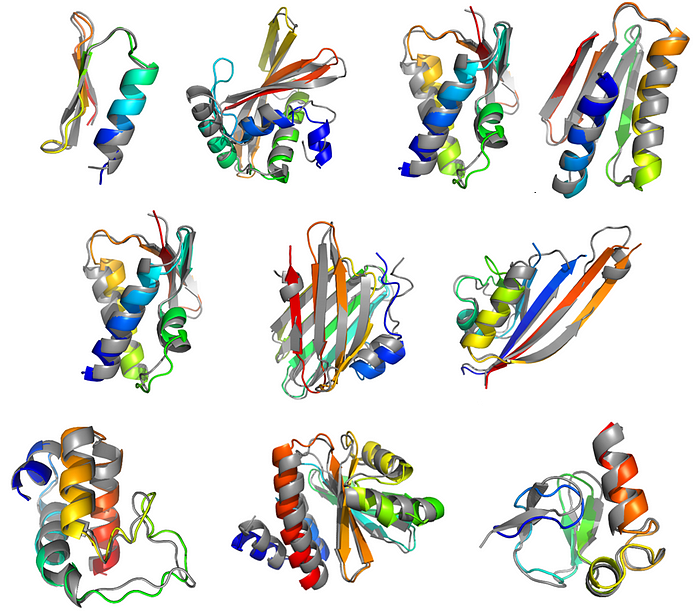
If DeepMind kind of “cracked” the problem of protein structure determination with its AlphaFold program, what is the next frontier in molecular modeling of proteins and biological systems? Here we have summarized the key takeaways from CASP15, the latest edition of the Critical Assessment of Structure Prediction competition from which AlphaFold 2 came out.
AlphaFold 2, introduced at CASP14 in December 2020, marked a huge leap forward in protein structure prediction. The release of AlphaFold 2’s open-source code in June 2021 triggered a surge of activity in the scientific community, leading to a revolution in the field of protein structure prediction. Researchers in molecular modeling and structural biology quickly embraced AlphaFold and its open-source code, tools like ColabFold and MMseqs2 made the technology accessible to a wide audience without barriers, novel structural databases started to be developed (see our previous post), and new applications were sought. For example, right after its launch researchers tweaked AlphaFold to model not just single proteins but their complexes too, and then DeepMind developed AlphaFold2-Multimer, designed to to better predict structures of multiprotein assemblies.
More globally, the new AI concepts introduced by Deepmind were reused to create new models for predicting protein structure and function, and even to redesign full proteins from scratch. The latest works, out just weeks before writing this blog entry, can model and design not only proteins but also their complexes with any other type of molecule -stay tuned for our upcoming article on them.
CASP15 and the Future of Modeling the Structures of Biological Systems
CASP15, held two years after AlphaFold 2’s groundbreaking debut at CASP14, witnessed a widespread adoption of AlphaFold in the scientific community. Moreover, several groups showcased significant improvements for individual proteins and protein assemblies over the basal AlphaFold 2 predictions, by tweaking inputs and sampling protocols.
Five of these groups stood out for their achievements. They used various strategies, including more efficient use of templates (structures presumably similar to the targets to be modeled), enhanced multiple sequence alignments, and custom modifications to AlphaFold. These innovations enabled them to produce better predictions, especially for the more complex protein structures.
New Categories in CASP15
CASP15 introduced four new prediction categories to address the evolving challenges in biomolecular modeling: modeling of RNA structure, of ligand-protein complexes, of large oligomeric structures, and of multiple alternative conformations. These categories reflect the changing landscape of molecular modeling and the need to address specific problems in the field, especially that of accounting for more than single, static protein molecules. Just 4 years ago the forefront was in modeling protein tertiary structure; but with AlphaFold 2, this problem was overcome and new levels of complexity were unlocked.
Enhancing Protein Assembly Prediction
CASP15 witnessed remarkable improvements in protein assembly prediction, with a 90% success rate in overall fold and interface contact predictions. This significant achievement was attributed to the incorporation of DeepMind’s AlphaFold2-Multimer approach into custom-built prediction pipelines. Customized multiple sequence alignments, optimized sampling, and manual assembly of subcomplexes were among the key factors contributing to this success.
Challenges still persist, especially in predicting structures with weak evolutionary signals and in modeling complexes that are way too large for current computer memory capacity.
Modeling Protein-Ligand Complexes
In the new category focused on ligand prediction, participants had to reproduce not just a protein’s structure but also how the small molecules (ligands, inhibitors, etc.) bound to it. As input, they were given protein sequences, nucleic acid sequences if present, SMILES line notation for ligands, and stoichiometries.
While small ions and rigid ligands were predicted with high fidelity, flexible ligands presented a more significant challenge. Usually the right pockets were found, but the ligand conformations and the exact protein-ligand contacts were off. Plenty of space for new work here -but stay tuned for what two just published methods can do.
Breaking the Conformational Ensemble Barrier
For the first time, CASP15 included a section on computing multiple conformations. The success in this track was variable, marking a promising advancement. For protein targets, enhanced sampling using variations of the AlphaFold 2 proved highly effective in generating multiple conformations. However, challenges remain in handling sparse or low-resolution experimental data and modeling RNA-protein complexes.
What’s in CASP15 for you at Nexco
As the field evolves at such fast pace, it is hard to keep track of new developments. But through contact with researchers active in molecular modeling and with our consulting experts who were CASP assessors in recent editions, we stay at the forefront.
To know more about us and learn how we can put the latest molecular modeling and other bioinformatics tools to work for you, check our website at https://www.nexco.ch/.
References
https://www.sciencedirect.com/science/article/pii/S0959440X23000684
https://onlinelibrary.wiley.com/doi/10.1002/prot.26515?af=R
https://onlinelibrary.wiley.com/doi/full/10.1002/prot.26598
https://onlinelibrary.wiley.com/doi/full/10.1002/prot.26601
Related Posts

Our location
Nexco Analytics Bâtiment Alanine Route de la Corniche 5B 1066 Epalinges, Switzerland
Give us a call
+41 76 509 73 73
Leave us a message
contact@nexco.chDo not hesitate to contact us
We will answer you shortly with the optimal solution to answer your needs
© 2024 Nexco Analytics. All Rights Reserved.